Publié le 14 juin 2016Lecture 6 min
Une homocystinurie B6 sensible
V. RIGALLEAU*, C. CORDROC'H*, C. GED**, REDONNET-VERNHET** *Service d'endocrinologie-nutrition **Laboratoire de Biochimie, CHU de Bordeaux
Monsieur C. est âgé de 42 ans lorsqu'il est hospitalisé en 2009 pour une hémiparésie gauche, conduisant à découvrir une thrombose de l'artère carotide interne droite.
Cas clinique
Monsieur C. est âgé de 42 ans lorsqu'il est hospitalisé en 2009 pour une hémiparésie gauche, conduisant à découvrir une thrombose de l'artère carotide interne droite. Son frère a présenté un infarctus du myocarde à l'âge de 50 ans. Ses antécédents personnels comportent un retard mental modéré, une hypertension artérielle et un tabagisme évalué à 5 années-tabac.
La recherche d'une cause à l'accident vasculaire cérébral précoce met en évidence :
- une cardiomyopathie hypokinétique, sans trouble du rythme ;
- une glycémie à jeun à 6 mmol/l, un bilan lipidique normal ;
- l'absence de localisation athéromateuse à l'examen écho-Doppler artériel des membres inférieurs.
Un dosage d'homocystéinémie totale plasmatique est réalisé : 225 μmol/l (N < 16,5).
Sur la chromatographie des acides aminés, la méthioninémie est élevée : 41 μmol/l (N < 29 μmol/l). Les taux sériques d'acide folique et de vitamine B12 sont normaux, la vitamine B6 sérique abaissée à 9,4 nmol/l (N 20-90). L’analyse du gène CBS codant la cystathionine bêta-synthase identifie une mutation faux sens fréquente associée aux formes B6-sensibles : le variant ile278thr, présent à l’état homozygote.
Un traitement par vitamine B6 est débuté : Bécilan® 250 mg x 2/j. L'évolution biologique des mois suivants est décrite sur le tableau.
L'homocystinurie congénitale
L'homocystinurie congénitale est une maladie héréditaire du métabolisme, de transmission autosomale récessive, liée à un déficit enzymatique en cystathionine bêta-synthase (CBS). Les sujets atteints ont une activité CBS effondrée ; ils sont homozygotes ou hétérozygotes composites pour un défaut du gène CBS, situé sur le chromosome 21(1). Dans la moitié des cas, l’activité CBS peut être restaurée par un traitement par son cofacteur, la vitamine B6. Le pronostic est alors nettement meilleur. Les sujets hétérozygotes ont une activité abaissée, mais qui suffit à maintenir une homocystéinémie normale, susceptible de s'élever après une charge orale en méthionine.
Il s’agit d’une maladie rare : les études pilotes de dépistage néonatal systématique par dosage de méthioninémie ont trouvé 145 cas pour 49 886 727 sujets(2), soit 1/345 000, ce qui correspondrait à 1 ou 2 nouveaux cas par an en France. Mais les formes B6-sensibles, de révélation plus tardive, échappent à ce dépistage. La recherche systématique de la mutation prévalente (c.833 > C p.Ile278Thr) associée aux formes B6 sensibles, chez 500 nouveau-nés danois, a trouvé 1,4 % d'hétérozygotes, ce qui correspondrait à 270 homozygotes sur l'ensemble de la population danoise ; or, seulement 10 cas étaient connus dans les centres spécialisés au Danemark(3). De nombreux cas sont donc probablement ignorés.
Mécanisme biochimique
Les conséquences métaboliques du déficit en CBS sont schématisées par la figure 1. Normalement, l'homocystéine est issue du métabolisme de la méthionine, acide aminé soufré indispensable. Le taux d'homocystéine peut s'élever dans deux cas, distingués par le dosage de méthioninémie sur la chromatographie des acides aminés :
- la méthioninémie est basse lorsque la reméthylation de l'homocystéine en méthionine est défectueuse : déficit en méthionine synthase, en MTHFR (5,10-méthylène-tétrahydro-folate-réductase), variants cobalamine C/D ;
- la méthioninémie est élevée lorsque la CBS est défectueuse, comme chez notre patient. Le déficit en CBS a deux conséquences :
- en amont, l'accumulation d'homo cystéine, qui a une toxicité vasculaire (artérielle et veineuse) et de son précurseur, la méthionine ;
- en aval, un déficit en cystine qui expliquerait les anomalies du tissu conjonctif, du tissu osseux et du cristallin.
L'ensemble aboutit à une maladie systémique, avec des manifestations thromboemboliques, mais aussi neurologiques, osseuses et ophtalmologiques, particulièrement dans les formes B6-résistantes.
Figure 1. Métabolisme de l’homocystéine.
Une maladie systémique
Les accidents thromboemboliques surviennent à tout âge : 25 % des sujets en ont présenté un à 20 ans, 50 % à 30 ans. Ils sont graves, à prédominance veineuse avec 50 % de phlébites et embolies pulmonaires, mais aussi 30 % d'accidents vasculaires cérébraux ; les accidents artériels périphériques (10 %) et les infarctus du myocarde (5 %) sont plus rares. Ils sont évitables, le risque passe de 1/25 années-patients à 1/250 sous traitement. Ils surviennent souvent au décours d'interventions chirurgicales, d'épisodes de déshydratation, lors de la grossesse, en post-partum ou sous contraception orale. Ils réduisent l'espérance de vie ; la mortalité est de 23 % à l'âge de 30 ans (4 % pour les formes B6- sensibles).
Les manifestations neurologiques sont variées : comitialité, syndromes extrapyramidaux, troubles de la personnalité et de l'humeur, mais dominées par le retard mental. Le quotient intellectuel moyen est à 64 (78 chez les B6-sensibles), mais une proportion notable de patients a un niveau intellectuel normal, une insertion sociale et professionnelle satisfaisante.
L'atteinte ophtalmologique est dominée par l'ectopie du cristallin, survenue pendant l'enfance en général, et liée à un défaut de polymérisation de la fibrilline au niveau de la zonule de Zinn. Bien que très évocatrice, elle n'est pas spécifique, pouvant aussi être observée au cours du syndrome de Marfan. Cette anomalie est gênante, dangereuse, mais curable. D'autres complications ophtalmologiques sont possibles : myopies sévères, rétinopathies, atrophies optiques, anomalies motrices oculo-palpébrales. Le suivi ophtalmologique est indispensable.
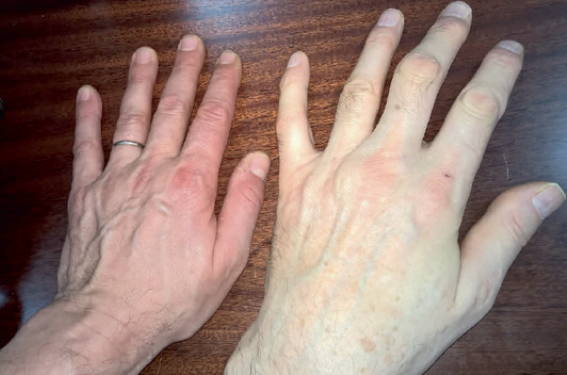
Les manifestations squelettiques donnent aux patients un aspect marfanoïde souvent caractéristique : dolichosténomélie, arachnodactylie (figure 2, concernant un patient B6-résistant), Pectus excavatum ou Pectus carinatum, scolioses aggravées par une ostéoporose généralement présente dès l'âge de 20 ans, qui justifie un traitement par biphosphonates.
Figure 2. Arachnodactylie chez un patient porteur d’une homocystinurie B6-non répondeur.
Diagnostiquer l'homocystinurie, c'est doser l'homocystéinémie
Il est impératif de doser l’homocystéinémie en cas d'accident vasculaire inexpliqué (figure 3).
Figure 3. Quand doser l’homocystéinémie ?
En cas d'hyperhomocystéinémie sévère (> 100 μmol/l), la démarche diagnostique comporte :
- un dosage de méthioninémie. S'il est élevé, c'est un déficit en CBS et on commence le traitement par vitamine B6 dans l'espoir que le patient y soit répondeur. S'il est bas, c'est une anomalie de la reméthylation ;
- un diagnostic génétique.
Les hyperhomocystéinémies modérées (15-30 μmol/l) sont en général secondaires à des causes acquises, aggravées par un déficit fonctionnel de la voie MTHFR (variant thermolabile présent dans 30 % de la population générale) en cas de taux intermédiaires (30-100 μmol/l). Ces causes sont : l'insuffisance rénale, les maladies intestinales, l'hypothyroïdie, l'hypogonadisme féminin, le psoriasis, les leucémies, la prise de certains médicaments (méthotrexate, phénytoïne). Un dépistage réalisé chez 18 000 Hollandais adultes (Hordaland Homocystein Study) a trouvé 67 sujets > 40 mmol/l (0,37 %), dont un sujet atteint d’homocystinurie par déficit en CBS confirmé(4) : 80 % avaient un déficit en acide folique, 20 % un déficit en vitamine B12, et 73 % étaient porteurs du variant prédisposant MTHFR. Leur hyperhomocystéinémie tendait à persister au long cours, mais elle s'est améliorée sous traitement par acide folique.
Le traitement par vitamine B6 (pyridoxine)
La vitamine B6 est un produit naturellement présent dans l'alimentation (viandes, œufs, choux, etc.), avec un apport nutritionnel conseillé de 1,5 à 2 mg/j. Les apports alimentaires de vitamine B6, et son taux sérique, ne sont pas un déterminant important du niveau de l'homocystéinémie dans la population générale. La carence observée chez notre patient est classique dans les déficits en CBS ; elle ne doit pas égarer le diagnostic car une carence en vitamine B6 ne donne jamais des taux aussi élevés d'homocystéinémie. Même avec un taux sérique normal, il fallait le traiter par de fortes doses de vitamine B6 (Bécilan® 500 mg/j).
Les patients B6-répondeurs normalisent en quelques semaines leur homocystéinémie avec ce traitement. Quelques patients ont une réponse partielle, qui peut nécessiter une dose encore plus forte (1 g/j, avec une supplémentation en acide folique associée), mais la plupart s'améliorent clairement, permettant éventuellement de réduire la dose de vitamine B6 à 100 mg/j au long cours. Les taux sériques de vitamine B6 sont nettement supérieurs à la normale (x 20 fois la normale) lors du traitement de l'homocystéinémie B6-sensible, justifiant une surveillance car l'intoxication à la vitamine B6 peut causer des neuropathies.
Les patients non répondeurs à la vitamine B6
Leur traitement est nettement plus difficile et spécialisé. Il repose sur :
- le régime pauvre en méthionine (donc hypoprotéique), supplémenté avec des substituts d'acides aminés sans méthionine (Maxamum X Met, HCU-Cooler), lors que la déficience mentale reste compatible avec l'éducation thérapeutique du patient ;
- la polyvitaminothérapie B6-B12-acide folique ;
- la bétaïne (Cystadane®, 100 mg/ kg/j) qui permet la reméthylation de l'homocystéine en méthionine.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :